Синдром Ангельмана у детей и взрослых
Существует ряд заболеваний, при которых выражения типа «береги себя и не заболеешь» звучат, по крайней мере, нелепо. Это патологии, при которых некоторые психические и физические отклонения заложены в организме ребенка еще до рождения, но вины родителей в этом нет. Такие заболевания вызываются мутациями или нарушениями в хромосомных наборах и называются хромосомными или генетическими. Синдром Ангельмана, синдромы Дауна, Патау, Эдвардса, Тернера, Прадера-Вилли – это лишь часть генетических заболеваний из довольно приличного списка.
Синдром счастливого человека
В этот раз мы поговорим о патологии, названной в честь педиатра из Англии Гарри Ангельмана, который впервые поднял вопрос об этой проблеме 1 965 году, столкнувшись накануне в своей практике с тремя необычными детьми, объединенными общими своеобразными симптомами. Врач назвал этих деток кукольными детьми и написал о них статью, которая изначально называлась «Дети-марионетки». Сама статья и ее название были написаны под впечатлением от картины, увиденной в одном из музеев Вероны. На картине был изображен смеющийся мальчик, и она носила название «Мальчик-марионетка». Ассоциация изображенного на картине ребенка с теми тремя детьми, с которыми Ангельман однажды столкнулся в своей практике, и подтолкнула педиатра объединить малышей в одну группу в связи с имеющимся у них заболеванием.
В том, что отмеченные в статье дети не были замечены другими врачами, нет ничего удивительного. Ведь на первый взгляд казалось, что у них совершенно разные заболевания, настолько отличалась общая клиническая картина болезни в 3 разных случаях. Может «новая» хромосомная патология и заинтересовала бы других ученых, но на то время генетика еще не была настолько развита, чтобы подтвердить гипотезу английского врача. Поэтому статья после определенного интереса к ней на долгое время была заброшена на дальнюю полку.
Следующее упоминание о синдроме Ангельмана, а именно так теперь называлась статья педиатра из Англии Г. Ангельмана, датируется началом 80-х годов ХХ столетия. И лишь в 1987 году удалось найти причину, по которой небольшая часть детей рождается с такими отклонениями, что со стороны они кажутся постоянно улыбающимися и счастливыми. На самом деле, это совсем не так, а улыбка – всего лишь гримаса, за которой скрывается несчастная человеческая душа и боль родителей.
Код по МКБ-10
Эпидемиология
Хромосомная мутация у ребенка, согласно статистике, может развиваться как на фоне подобных мутаций у родителей, так и при отсутствии таковых. Четкого наследственного характера у синдрома Ангельмана (СА) не прослеживается, но вероятность развития патологии у родителей с хромосомными мутациями достаточно высока.
Интересным является и то, что если в семье уже есть ребенок с СА, существует однопроцентная вероятность рождения второго такого же ребенка, даже если родители здоровы.
Точных статистических данных о количестве больных с синдромом Ангельмана все еще нет. Возможно, виной всему разнообразие симптомов, которые могут возникать в определенном составе или длительное время не возникать вообще. Предполагается, что распространенность болезни составляет: 1 ребенок на 20000 новорожденных. Но эта цифра очень приблизительна.
Причины синдрома Ангельмана
Синдром Ангельмана – медицинское название хромосомной патологии, но оно далеко не единственное. В народе эту болезнь называют и синдромом кукольных детей, и синдромом счастливой марионетки, и синдромом Петрушки, и синдромом смеющейся куклы. Да каких только названий не придумают люди (порой даже оскорбительных для самих пациентов и их родителей), но болезнь есть болезнь, как бы забавно это ни выглядело внешне и какими бы причинами ни было вызвано.
А причинами развития синдрома Ангельмана, как и многих других генетических патологий, во всех случаях становятся нарушения в строении одной из хромосом или хромосомного набора в целом. Вот только в нашем случае вся проблема заключается в 15 хромосоме, переданной от матери. Т.е. отцовская хромосома в данном случае не имеет никаких отклонений, а вот женская претерпевает определенные мутации.
По типу хромосомной аномалии синдром Ангельмана относится к хромосомным мутациям. Такими мутациями считаются:
- Делеция (отсутствие участка хромосомы, содержащего определенный набор генов; если отсутствует один из генов речь идет о микроделеции), являющаяся результатом двух разрывов и одного воссоединения, когда утрачивается участок исходной хромосомы.
- Дупликация (наличие лишнего участка в хромосоме, являющегося копией уже имеющегося), которая в большинстве случаев приводит к смерти человека, реже – к бесплодию.
- Инверсия (перевертывание одного из участков хромосомы на 180 градусов, т.е. в обратную сторону, и тогда гены в нем оказываются расположенными в обратном порядке), когда разорванные концы хромосомы соединяются в порядке, отличном от исходного.
- Инсерция (если часть генетического материала в хромосоме оказывается не на своем месте),
- транслокация (если некий участок хромосомы присоединяется к другой хромосоме; такая мутация может быть взаимной без потери участков).
Получая мутировавшую хромосому от ничего не подозревающей мамы ребенок заранее обречен родиться с отклонениями. Наиболее частой причиной развития синдрома Ангельмана считается все же делеция материнской 15 хромосомы, когда в ней отсутствует небольшой участок. Реже встречающимися мутациями при синдроме «смеющейся куклы» считаются:
- транслокация,
- одноотцовская дисомия (если ребенок получил пару хромосом от отца, материнская хромосома при этом отсутствует),
- мутация генов в составе ДНК, являющихся одновременно и основным строительным (генетическим) материалом и инструкцией к правильному его использованию (в частности мутация гена ube3a в материнской хромосоме).
Наличие одной из таких мутаций у родителей является фактором риска развития синдрома Ангельмана у детей. Но не только хромосомные мутации, но и геномные (которые связаны с количественным изменением в хромосомных наборах и встречаются чаще хромосомных) могут спровоцировать развитие болезни у ребенка. К распространенным геномным мутациям можно отнести трисомию хромосом (если у человека хромосомный набор имеет более 46 хромосом).
Чтобы патология появилась у малыша вовсе не обязательно, чтобы у родителей были хромосомные отклонения. И все же есть некий процент пациентов, заболевание у которых имеет наследственный характер.
Патогенез
Углубимся немного в биологию, точнее в генетику. Генетическая информация каждого отдельного человеческого организма заключена в 23 парах хромосом. Одна хромосома из пары передается ребенку от отца, другая – от матери. Все пары хромосом отличаются формой и размером и несут в себе определенную информацию. Так, 23 пара хромосом (Х и Y хромосомы) отвечает за формирование у малыша половых признаков (ХХ – девочка, ХY- мальчик, при этом Y-хромосому ребенок может получить лишь от отца).
В идеале ребенок получает от родителей 46 хромосом, которые и формируют его генетические признаки, предопределяя его как индивида. Большее число хромосом называется трисомией и считается отклонением от нормы. Например, наличие 47 хромосомы в хромосомном наборе (кариотипе, определяющем видовые и индивидуальные признаки) вызывает возникновение синдрома Дауна.
Если хромосомы подкрасить специальным красителем, то в микроскопе можно увидеть полосы разных оттенков вдоль каждой из них. Внутри каждой полосы находится огромное число генов. Все эти полосы учеными пронумерованы и имеют фиксированное расположение. Отсутствие одной из полос считается отклонением от нормы. При синдроме Ангельмана можно очень часто наблюдать отсутствие сегментов материнской хромосомы в промежутке q11-q13, расположенных в длинном плече, количество оснований ДНК в которых всего-навсего порядка 4 миллионов.
Основной составляющей хромосомы считается невероятно длинная молекула ДНК, содержащая тысячи генов и десятки и сотни миллионов азотистых оснований. Так, 15 хромосома, ответственная за развитие синдрома Ангельмана и еще нескольких других, содержит 1200 генов и порядка 100 млн. оснований. Любые нарушения в строении молекулы ДНК обязательно отразятся на внешности и развитии будущего ребенка.
Генетическая информация, содержащаяся в генах, преобразуется в белок или РНК. Этот процесс называют экспрессией генов. Таким образом полученная от родителей генетическая информация получает и форму, и содержание, воплощающиеся в их неповторимом наследнике женского или мужского пола.
Существует ряд патологий с неклассическим типом наследования, включая и синдром Ангельмана, при которых полученные от родителей гены в составе парных хромосом несут своеобразный отпечаток родителей и проявляют себя по-разному.
Так вот, синдром Ангельмана является ярким примером геномного импринтинга, при котором экспрессия генов в организме ребенка находится в прямой зависимости от того, от кого из родителей получены аллели (разные формы одного гена, полученные от отца и матери, расположенные на идентичных участках парных хромосом). Т.е. к возникновению синдрома приводят лишь аномалии в материнской хромосоме, в то время как мутации и нарушения структуры отцовской хромосомы вызывают совершенно иные патологии.
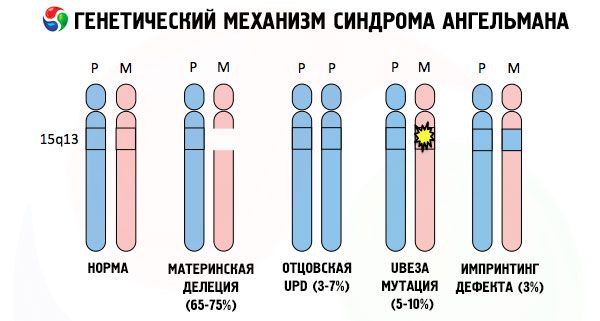
При данной патологии имеет место отсутствие определенных генов в материнской хромосоме или же потеря/снижение активности отдельных генов (в подавляющем большинстве случаев гена ube3a, участвующего в метаболизме убиквитина – белка, регулирующего деградацию других белков). Вследствие этого у ребенка диагностируются отклонения в психическом развитии и физические уродства.
Симптомы синдрома Ангельмана
Симптоматика синдрома Ангельмана затрагивает различные стороны жизни и развития ребенка: физическую, неврологическую, психическую. На основании этого можно выделить 3 группы симптомов, указывающих на развитие данной патологии.
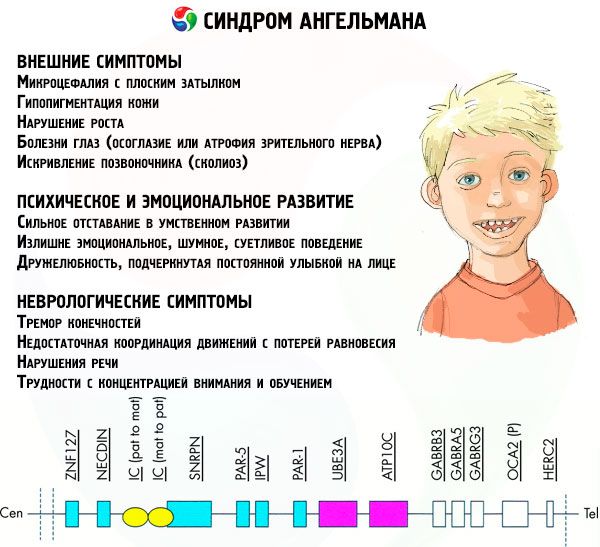
- Внешние или физические симптомы:
- непропорционально маленькая голова по сравнению с туловищем и конечностями, имеющими нормальный размер,
- слишком широкий рот,
- на лице практически всегда присутствует улыбка (с открытым ртом),
- редкие зубы,
- узкая верхняя губа,
- часто высунутый широкий язык,
- выступающая нижняя челюсть,
- острый подбородок,
- очень светлая кожа, часто и волосы (альбинизм, связанный с тем, что в организме не вырабатывается пигмент меланин),
- темные пятна на светлой коже (гипопигментация из-за недостаточной выработки меланина)
- физические или внешние симптомы: болезни глаз, такие как косоглазие или атрофия зрительного нерва,
- искривление позвоночника (сколиоз),
- негнущиеся ноги (при ходьбе человек не сгибает ноги в коленях из-за малой подвижности суставов, отсюда сравнение с кукольной походкой).
- Симптомы, связанные с психическим и эмоциональным развитием:
- сильное отставание в умственном развитии,
- излишне эмоциональное, шумное, суетливое поведение,
- частое хлопанье в ладоши,
- выраженная дружелюбность, подчеркнутая постоянной улыбкой на лице,
- частый беспричинный смех.
- Неврологические симптомы:
- тремор конечностей,
- недостаточная координация движений с потерей равновесия,
- пониженный тонус мышц,
- разнообразные нарушения сна,
- частые истерические припадки в детском возрасте,
- нарушения речи (ребенок поздно начинает говорить, у него плохие коммуникационные способности и невнятная речь),
- гиперактивность на фоне повышенной возбудимости,
- трудности с концентрацией внимания и обучением.
Но это все обобщенная картина болезни. На самом деле клиническая картина синдрома Ангельмана в большой степени зависит от стадии развития болезни и вида хромосомной мутации, вызвавшей патологию. А это значит, что у разных пациентов симптоматика болезни может существенно отличаться, что долгое время не позволяло выделить патологию среди других со сходной клинической картиной.
Среди общего числа симптомов можно выделить такие, которые характерны всем без исключения пациентам:
- тяжелые отклонения в умственном развитии,
- неадекватное поведение (беспричинный смех, повышенная возбудимость, слабая концентрация внимания, состояние эйфории),
- недоразвитие моторики,
- плохая координация движений, атаксия ходьбы (неравномерный темп, раскачивание из стороны в сторону и т.п.), тремор конечностей.
- нарушение речевого развития с преобладанием невербальных средств общения.
Среди симптомов, встречающихся у подавляющего большинства пациентов, можно выделить такие:
- непропорциональность головы и туловища, вызванная задержкой физического развития,
- у многих пациентов форма черепа такова, что размер головного мозга остается меньше, чем у здоровых людей (микроцефалия),
- эпилептические припадки в возрасте до 3 лет с поступательным уменьшением силы и частоты в старшем возрасте,
- искажение показателей ЭЭГ (колебания и высокая амплитуда волн низкой частоты).
Эти симптомы встречаются довольно часто, тем не менее у 20% пациентов с синдромом Ангельмана они отсутствуют.
Еще реже можно диагностировать такие проявления болезни как:
- выраженное или легкое косоглазие,
- слабый контроль за движением языка, в результате чего пациенты часто без причины высовывают язык,
- трудности с глотанием и сосанием, особенно в младшем детской возрасте,
- нарушение пигментации кожи и глаз,
- поднятые или согнутые во время ходьбы руки,
- гиперрефлексия,
- нарушения сна, особенно в детстве,
- частое слюнотечение,
- неуемная жажда,
- излишне активные жевательные движения,
- гиперчувствительность к теплу,
- плоский затылок,
- выдвинутая вперед нижняя челюсть,
- гладкие ладони.
Довольно большой процент пациентов имеет проблемы с мочеиспусканием, которое они плохо контролируют, нарушение мелкой моторики, что создает трудности в самообслуживании и обучении, лишний вес. Практически у всех пациентов половое созревание начинается позже, чем у здоровых сверстников.
Дети с синдромом Ангельмана хорошо воспринимают устную речь и понимают ее, но участвовать в разговоре не желают, ограничивая свою речь несколькими десятками слов, необходимых в быту. Зато во взрослом возрасте такие пациенты выглядят моложе ровесников без генетических патологий.
Многие симптомы синдрома Ангельмана непостоянны, поэтому с возрастом клиническая картина болезни значительно изменяется. Судороги и эпилептические приступы становятся более редкими или исчезают вообще, пациент становится менее возбудимым, налаживается сон.
Осложнения и последствия
Синдром Ангельмана – тяжелая, практически неизлечимая на сегодняшний день хромосомная патология, которая лишает пациентов возможности жить нормальной жизнью. То, какой будет жизнь малыша с СА, в большой степени зависит от вида хромосомной аномалии.
Дублирование участка хромосомы в большинстве случаев является несовместимым с жизнью. И если даже такие пациенты не умирают в младенчестве и достигают половой зрелости, возможность иметь детей у них отсутствует.
Делеция или отсутствие части генов, которая встречается при синдроме Ангельмана чаще всего, является препятствием к тому, чтобы ребенок научился ходить и говорить. У таких детей умственная отсталость представлена в более тяжелой форме, чаще случаются приступы эпилепсии, при этом их интенсивность значительно сильнее, чем у пациентов с иными хромосомными аномалиями.
Если же имеет место лишь мутация одного гена, при должном внимании и подходе ребенка можно научить основам самообслуживания, коммуникации и общению в коллективе, хотя он все равно будет отставать в развитии от своих сверстников.
Для детей с синдромом Ангельмана, доброжелательных по натуре, основным является любовь и внимание родителей. Лишь в таком случае обучение ребенка будет давать плоды, пусть даже небольшие. Конечно, обучаться в обычной школе пациенты с СА не смогут. Им нужны специальные занятия, где детей сначала научат концентрировать внимание, а затем уже понемногу будут давать основы школьных знаний.
Диагностика синдрома Ангельмана
Синдром Ангельмана – врожденная патология развития. Но в силу некоторых обстоятельств диагностировать ее в младенчестве и раннем детском возрасте чаще всего не представляется возможным. Виной тому неспецифичность и слабая выраженность симптомов у грудничков и малышей до 3 лет. Да и распространенность болезни у нас в стране не столь велика, чтобы врачи научились распознавать ее среди ей подобных.
Синдром Ангельмана у детей грудного возраста может проявляться в виде пониженного тонуса мышц, что проявляется в виде проблем с питанием (слабость сосательного и глотательного рефлекса), а позже трудностями в обучении хождению (такие дети значительно позже начинают ходить). Эти симптомы являются первыми признаками отклонения в развитии малыша, которое вполне может быть связано с хромосомной аномалией. Подтвердить это предположение может лишь генетический анализ.
Особое внимание уделяется деткам, у родителей которых имеются различные геномные или хромосомные нарушения. Ведь болезнь попервах может никак себя не проявлять, и если выявить патологию вовремя, начав усиленно заниматься с ребенком, можно достигнуть значительно больших успехов в обучении, затормозив прогрессирование болезни.
Если у родителей имеются различные хромосомные аномалии, генетический анализ проводят еще до рождения малыша, поскольку СА является одной из патологий, которые можно выявить еще в зачаточном состоянии.
Забор материала для генетического исследования при этом можно провести двумя путями:
- инвазивным (с определенным процентом риска, поскольку требуется проникнуть в матку для того, чтобы взять пробу околоплодной жидкости),
- неинвазивным (анализ ДНК младенца по крови матери).
Далее проводятся следующие исследования:
- флуоресцентная in situ гибридизация (FISH-метод) – связывание ДНК-зонда, помеченного специальным красителем, с исследуемой ДНК с последующим исследованием под микроскопом.
- анализ мутации в гене ube3a и импринтинговых генах,
- анализ метилирования ДНК при помощи специальных методов, применяемых в генетике.
Генетические анализы дают достаточно точные сведения в случае хромосомных отклонений, а значит будущие родители заранее знают к чему им готовиться. Тем не менее, бывают и исключения. У определенной группы пациентов при наличии всех указывающих на патологию симптомов результаты анализов остаются в норме. Т.е. выявить патологию можно лишь внимательно наблюдая за ребенком с самого раннего детства: как кушает, когда начал ходить и говорить, сгибает ли ножки при ходьбе м т.д.
Помимо FISH-метода, среди методов инструментальной диагностики при синдроме Ангельмана можно выделить томографию (КТ или МРТ) , помогающую определить состояние и размеры головного мозга, и электроэнцефалограмму (ЭЭГ), показывающую как работают отдельные отделы головного мозга.
Окончательный диагноз врачи устанавливают обычно в возрасте 3-7 лет, когда у пациента уже налицо большинство симптомов и видна динамика развития болезни.
Дифференциальная диагностика
Синдром Ангельмана – генетическая патология, фактически не имеющая специфических проявлений. Большинство симптомов в равной степени могут указывать как на СА, так и на другие генетические патологии.
Дифференциальная диагностика при синдроме Ангельмана проводится со следующими патологиями:
- Синдром Питта –Хопкинса (пациенты характеризуются отставанием в умственном развитии, веселым характером, улыбчивостью, у них довольно большой и широкий рот, отмечается микроцефалия). Отличие – приступы гипервентиляции и задержки дыхания в состоянии бодрствования.
- Синдром Кристиансона (пациенты умственно отсталые люди с веселым нравом, не умеющие разговаривать, им характерны микроцефалия, атаксия, судороги, непроизвольные движения мышц).
- Синдром Моуота- Вильсон (симптомы: умственная отсталость, эпилептические припадки, острый подбородок, открытый рот, выражение счастья на лице, микроцефалия). Отличие – большое расстояние между глазами, глаза скошены вовнутрь, кончик носа закруглен, ушная раковина повернута назад.
- Синдром Кабуки (характерны легкая и средняя степень умственной отсталости, проблемы с речью и моторикой, слабость мышц, эпилептические припадки, микроцефалия, большие промежутки между зудами, нарушения координации). Отличие – брови в виде арки, вывернутая латеральная часть нижнего века, широко посаженные глаза, длинные глазные щели с длинными густыми ресницами.
- Синдром Ретта (дифференциация с СА у женщин). Симптомы: задержка речевого развития, судорожные припадки, микроцефалия. Отличие – нет счастливого выражения на лице, бывают приступы апноэ и апраксия, которая со временем прогрессирует.
- Синдром аутосомно-рецессивной ментальной тардации 38 (симптомы: заметная умственная отсталость с задержкой в развитии моторных навыков и речи, слабость мышц, проблемы с кормлением в грудном возрасте, импульсивность). Отличие – голубой цвет радужной оболочки глаза.
- Синдром дупликации гена МЕСР 2 (дифференциация с СА у мужчин). Симптомы: тяжелая умственная отсталость, слабость мышц с детства, проблемы с речью либо ее отсутствие, эпилепсия. Отличия – прогрессирующая миопатия, постоянно повторяющиеся инфекции.
- Синдром Клифстра (симптоматика: проблемы с речью и мышлением, мышечная слабость, нарушения сна, недостаток внимания, приоткрытый рот, гиперактивность, судороги, атаксия, нарушения равновесия). Отличия – плоское лицо, короткий курносый нос, широко посаженные глаза, большая вывернутая нижняя губа, приступы агрессии.
- Синдром Смит-Магенис (характерны судороги, проблемы со сном, нарушения интеллектуального и моторного развития). Отличия – широкое и плоское лицо, выпуклость лба.
- Синдром Кулена- де Фриза (легкая и средняя умственная отсталость, слабость мышц, судорожные приступы, дружелюбие). Отличия – длинное лицо с высоким лбом, оттопыренные уши, косой разрез глаз, большая подвижность суставов, врожденные патологии сердца.
- Синдром Филан – Мак-Дермид (симптомы: умственная отсталость, нарушения речи или ее отсутствие). Отличия – большие руки с развитыми мышцами, слабость мышц с самого рождения, слабое потоотделение.
Сходными с синдромом Ангельмана симптомами могут «похвастать» и такие патологии как дефицит аденилсукциназы, синдром аутосомно-рецессивной ментальной ретардации 1, синдром дупликации хромосомы 2q23.1, синдромы гаплонедостаточности генов FOXG1, STXBP1 или MEF2C и некоторые другие.
Задача врача - поставить точный диагноз, дифференцировав синдром Ангельмана от патологий со сходной симптоматикой, и назначить эффективное лечение, актуальное для диагностированной степени развития болезни.
Лечение синдрома Ангельмана
Синдром Ангельмана относится к разряду тех патологий, поиском эффективного лечения которых медицина занимается и по сей день. Этиологическое лечение заболевания находится в стадии разработки различных методов и средств, многие из которых еще не прошли тестирование на людях. А значит пока что врачам приходится ограничиваться симптоматической терапией, помогающей хоть как-то облегчить незавидное положение детей и взрослых с синдромом марионетки, страдающих от эпилептических припадков, слюнотечения, гипотонии и нарушений сна.
Так снизить частоту и силу эпилептических приступов можно при помощи правильно подобранного противосудорожного препарата. Но вся сложность в том, что приступы у больных с СА отличаются от обычных эпилептических приступов тем, что им характерны несколько видов судорог, а значит, облегчить состояние можно будет введением сразу нескольких препаратов.
Наиболее популярными противосудорожными препаратами, применяемыми для лечения синдрома Ангельмана, считаются: вальпроевая кислота, топирамат, ламотриджин, леветирацетам, клоназепам и препараты на их основе. Реже применяются лекарства на основе кармазепина, фенитоина, фенобарбитала, этосуксимида, поскольку некоторые из них могут спровоцировать парадоксальный эффект, заключающийся в усилении и увеличении частоты эпилептических припадков. Такое случается если препарат используется в составе монотерапии.
Для лечения слюнотечения обычно применяют 2 метода: лекарственный (препараты подавляющие образование слюны) и оперативный, заключающийся в реимплантации слюнных протоков. Но в случае СА эти методы считаются неэффективными, и вопрос по-прежнему остается открытым. Родителям и тем, кто ухаживает за такими пациентами, приходится уделять этому моменту особое внимание, поскольку сами больные обычно не контролируют слюнотечение, а некоторые попросту не в состоянии позаботиться о себе.
Еще одна проблема – малая продолжительность сна. Зачастую дети с синдромом Ангельмана спят не более 5 часов, что негативно сказывается на работе всего организма. Легковозбудимые, активные детки, любящие игры и общение (пусть даже они и стараются ограничиться невербальными способами), заметно устают за день. Чтобы хорошо отдохнуть, организму нужен крепкий полноценный сон, но вот с этим-то как раз и загвоздка.
Казалось бы, для улучшения сна у легковозбудимых пациентов должно быть достаточно препаратов с седативным эффектом (фенотиазинов и атипичных антипсихотиков), успокаивающих нервную систему. Но в случае СА применение таких средств чревато возникновением негативных эффектов. Поэтому врачи отдают предпочтение все же легким снотворным препаратам, таким как «Мелатонин» (натуральный гормональный препарат на основе гормона сна), который дают пациентам за час до отхода ко сну в количестве 1 таблетки, и «Дифенгидрамин». кратность приема и дозировка которого устанавливается врачом в зависимости от состояния и возраста пациента.
Иногда у больных с синдромом Ангельмана наблюдаются проблемы с пищеварением и стулом. Наладить стул можно при помощи слабительных препаратов (лучше растительного происхождения).
А можно подойти к проблеме иначе, как это сделали американские медики, основываясь на некоторых методиках лечения аутизма, ведь многие симптомы, свойственные СА, характерны и для аутизма (импульсивность, непроизвольные движения, повторяющиеся действия, нарушение внимания, проблемы в общении и т.д.). Было замечено, что введение гормона секретина, нормализующего пищеварение и стул, положительно влияет и на внимание пациентов, а окситоцин помогает улучшить познавательные способности ребенка и память, скорректировать поведение.
Правда, одними гормонами тут не обойтись, особенно если речь заходит о детях. При синдроме Ангельмана показана поведенческая терапия, работа с психологом и логопедом (обучение невербальным способам общения и языку жестов). Обучение таких деток должно строиться по индивидуальной программе с участием специально обученных преподавателей, психолога и родителей. К сожалению, такое возможно не везде, и семьи остаются со своей проблемой наедине.
Поскольку многие маленькие пациенты с СА страдают от низкого мышечного тонуса и проблем с суставами, большое внимание уделяется физиотерапевтическому лечению. Чаще всего врачи прибегают к использованию парафиновых аппликаций, электофорезу, магнитотерапии.
Активный тонизирующий массаж и специальные упражнения лечебной физкультуры помогут больному малышу спустя время уверенно стоять на ногах и ходить. Особенно полезна в этом плане аквагимнастика, которую при СА рекомендуется проводить в прохладной воде. Она повышает тонус мышц и учит малыша владеть своим телом, координировать движения.
Противосудорожное лечение
Наиболее опасным симптомом при синдроме Ангельмана являются судорожные приступы, сходные с приступами при эпилепсии. Этот симптом наблюдается у 80% пациентов, а значит нужно всем им назначить эффективное противосудорожное лечение.
Лечение эпилептических приступов проводится при помощи витаминов и противосудорожных препаратов. При синдроме Ангельмана, сопровождающемся судорожным синдромом, полезными окажутся витамины группы В, а также витамины С, Д и Е. Но назначать витаминотерапию самостоятельно в данном случае очень опасно, ведь бесконтрольный прием витаминов может снизить эффективность противоэпилептических препаратов и спровоцировать новые, более тяжелые и продолжительные приступы.
Подбором противосудорожных лекарств и назначением их эффективной дозировки также должен заниматься врач-специалист. Он же решает, достаточно ли будет одного препарата или же пациенту придется длительное время принимать 2 и более лекарства.
Большинству пациентов врачи назначают препараты вальпроевой кислоты («Вальпроевая кислота», «Депакин», «Конвулекс», «Вальпарин» и др.), которые предупреждают судороги, улучшают настроение и психическое состояние больных.
Вальпроевая кислота выпускается в виде таблеток, сиропа и инъекционных растворов. Наиболее популярным препаратом считается лекарство пролонгированного действия «Депакин» в таблетках и в виде раствора для внутривенного введения. Дозировка препарата определяется врачом индивидуально в зависимости от веса, возраста и состояния пациента.
Принимают препарат во время еды от 2 до 3 раз в день. Средняя суточная доза – 20-30 мг на 1 килограмм веса пациента, максимальная – 50 мг/кг в сутки.
Противопоказания к применению. Не применяется при нарушениях работы печени и поджелудочной железы, геморрагическом диатезе, гепатите, порфирии и гиперчувствительности к препарату.
Среди побочных эффектов можно выделить дрожание рук, нарушения пищеварения и стула, изменения массы тела.
«Топирамат» также является препаратом выбора при СА. Он производится в виде таблеток и применяется как в составе монотерапии, так и в комплексе с другими лекарствами.
Способ применения и дозировка. Принимают таблетки вовнутрь независимо от приема пищи. Начальная суточная доза для взрослых составляет 25-50 мг, для детей – 0,5-1 мг/кг. Каждую неделю дозировку увеличивают согласно назначениям врача.
Препарат нельзя принимать при беременности и лактации, а также при повышенной чувствительности к его компонентам. Лекарство имеет множество различных побочных эффектов.
Препараты, которые может назначить врач при синдроме Ангельмана: «Кломазепам», «Ривотрил», «Ламотриджин», «Сейзар», «Ламиктал», «Леветирацетам», «Кеппра», «Эпитерра» и др.
Народное лечение и гомеопатия
Средства народной медицины, как и гомеопатические препараты, конечно же, отличаются сравнительной безопасностью, но вот эффективность такого лечения относительно синдрома Ангельмана можно считать спорной.
Хотя кое в чем народное лечение все же может помочь. Речь идет о купировании эпилептических приступов. В этом плане лечение травами может оказаться довольно эффективным.
Хороший эффект оказывает лекарственный сбор на основе пиона, солодки и ряски (компоненты берут в равных количествах). Травы нужно перемолоть в муку. Спустя 2 недели от начала приема можно заметить значительное снижение частоты судорожных приступов.
Полезен при судорогах и отвар лаванды (1 ч.л. на стакан кипятка). Состав кипятят 5 минут и настаивают в течение получаса. Принимают лекарство на ночь в течение 14 дней.
Эффективным при эпилептических приступах считается водный (или же спиртовой) настой пустырника.
Из препаратов гомеопатии для предупреждения судорожных приступов при синдроме Ангельмана можно применять лекарства на основе ромашки и пустырника, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Но нужно учитывать, что эффективные и безопасные дозы препаратов в каждом конкретном случае может назначить лишь врач гомеопат.
Профилактика
Как читатель уже, вероятно, понял, предотвратить мутацию генов и другие хромосомные аномалии медицине пока не под силу, впрочем, как и исправить ситуацию. Такое может случиться с каждым, ведь дети с синдромом Ангельмана рождаются и у здоровых родителей, а генетика, которая на данный момент является одной из самых малоизученных отраслей медицины, не может пока этого объяснить.
Единственное, что можно сделать, это ответственно отнестись к планированию беременности, вовремя стать на учет и обследоваться. Но опять же, такая мера будет скорее не профилактическая, а познавательная, как и любые обследования. Зато молодые родители заранее будут знать, к чему готовиться, и в случае положительного ответа решат, смогут ли они взять на себя такую ответственность, как воспитание больного ребенка.
Прогноз
Прогноз при синдроме Ангельмана зависит от характера хромосомной аномалии и своевременности ее обнаружения. Тяжелее всего приходится тем деткам, чья 15 хромосома содержит «пропуски» генов (делеция). Вероятность ходить и разговаривать у таких пациентов чрезвычайно мала. Остальные случаи при внимательном подходе и любви к своему ребенку поддаются коррекции.
Стать полноценными членами общества такие больные, увы, не смогут, при всем том, что они далеко не глупы, понимают речь и ее смысл. Вот только проблемы с общением у них остаются на всю жизнь. Пациентов с детства можно обучить языку жестов, но нельзя заставить общаться при помощи слов. Лексикон «говорящих» больных ограничивается минимумом слов, употребляемых в быту (5-15 слов).
Что касается продолжительности жизни и общего состояния здоровья больных с синдромом Ангельмана, то здесь цифры колеблются на средних показателях. Во взрослой жизни пациенты в основном сталкиваются с такими проблемами со здоровьем, как сколиоз и ожирение, которые при правильном подходе к лечению не опасны для жизни.
Last reviewed: 25.06.2018