Медицинский эксперт статьи

Новые публикации
Болезнь Шарко-Мари-Тута
Последняя редакция: 23.11.2021

Весь контент Web2Health проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Перонеальная мышечная атрофия, синдром или болезнь Шарко-Мари-Тута является целой группой хронических наследственных заболеваний с повреждением периферических нервов.
По МКБ-10 в разделе болезней нервной системы код данного заболевания – G60.0 (наследственная моторная и сенсорная невропатия). Также оно внесено в перечень орфанных заболеваний.
Код по МКБ-10
Эпидемиология
Согласно данным клинической статистики, распространенность всех типов болезни Шарко-Мари-Тута на 100 тыс. населения составляет 19 случаев (по другим источникам, один случай на 2,5-10 тыс. населения).
На ШМТ 1 типа приходится около двух третей случаев (один случай на 5-7 тыс. населения), и почти 70% из них связаны с дублированием гена PMP22. В мире данным типом заболевания страдают более 1,2 млн. человек.
Частота случаев ШМТ 4 типа оценивается в 1-5 случаев на 10 тыс. детей. [1]
Причины болезни Шарко-Мари-Тута
Согласно классификации полинейропатических синдромов, перонеальная (малоберцовая) мышечная атрофия, невральная амиотрофия Шарко-Мари-Тута или болезнь Шарко-Мари-Тута (сокращенно – ШМТ) относится к генетически обусловленным моторно-сенсорным полинейропатиям. [2]
То есть причины ее возникновения – генетические мутации. И в зависимости от характера генетических отклонений различаются основные типы или виды данного синдрома: демиелинизирующий и аксональный. К первой группе относят болезнь Шарко-Мари-Тута 1 типа (ШМТ1), возникающую вследствие дублирования гена PMP22 на хромосоме 17, который кодирует трансмембранный периферический миелиновый белок 22. В результате происходит сегментарная демиелинизация оболочки аксонов (отростков нервных клеток) и снижение скорости проведения нервных сигналов. Кроме того, мутации могут быть в некоторых других генах.
Аксональной формой является болезнь Шарко-Мари-Тута 2 типа (ШМТ2), которая затрагивает сами аксоны и связана с патологическими изменениями гена MFN2 в локусе 1p36.22, кодирующего мембранный белок митофузин-2, необходимый для слияния митохондрий и формирования функциональных митохондриальных сетей внутри клеток периферических нервов. Различают более десятка подтипов ШМТ2 (с мутациями в конкретных генах).
Следует отметить, что в настоящее время выявлено более сотни генов, повреждение которых, передаваемое по наследству, вызывает различные подтипы болезни Шарко-Мари-Тута. Например, при мутациях гена RAB7 развивается ШМТ типа 2В; альтерация гена SH3TC2 (кодирующего один из белков мембран шванновских клеток) вызывает ШМТ типа 4C, которая проявляется в детском возрасте и характеризуется демиелинизацией моторных и сенсорных нейронов (различают полтора десятка форм 4 типа этой болезни).
Начинает развиваться в раннем детстве и редкая ШMT 3 типа (называемая синдромом Дежерина-Сотта), обусловленная мутациями генов PMP22, MPZ, EGR2 и др.
При возникающей в возрасте 5-12 лет ШМТ 5 типа отмечается не только моторная нейропатия (в виде спастического парапареза нижних конечностей), но и повреждение зрительного и слухового нервов.
Мышечная слабость и атрофия зрительного нерва (с потерей зрения), а также проблемы с равновесием характерны для ШМТ 6 типа. А при 7 типе болезни Шарко-Мари-Тута наблюдается не только моторно-сенсорная нейропатия, но и заболевание сетчатки в виде пигментного ретинита.
Более распространенная среди мужчин X-сцепленная ШMT или болезнь Шарко-Мари-Тута с тетрапарезом конечностей (ослаблением движения обеих рук и ног) – относится к демиелинизирующему типу и считается результатом мутации гена GJB1на длинном плече Х-хромосомы, который кодирует коннексин 32 – трансмембранный белок шванновских клеток и олигодендроцитов, регулирующий передачу нервных сигналов. [3]
Факторы риска
Основной фактор риска ШМТ – наличие данной болезни в семейном анамнезе, то есть у близких родственников.
Как утверждают генетики, если носителями аутосомно-рецессивного гена болезни Шарко-Мари-Тута являются оба родителя, риск рождения ребенка, у которого разовьется это заболевание, составляет 25%. А риск того, что ребенок будет носителем этого гена (но он сам не будет иметь никаких симптомов) оценивается в 50%.
В случае Х-сцепленного наследования (когда мутировавший ген находится на Х-хромосоме женщины) существует 50% риск того, что мать передаст этот ген сыну, и у него разовьется болезнь ШМТ. При рождении ребенка женского пола болезнь может не возникнуть, но унаследовать дефектный ген могут сыновья дочери (внуки) – с развитием заболевания.
Патогенез
При любых типах болезни Шарко-Мари-Тута ее патогенез обусловлен наследственной аномалией периферических нервов: моторных (двигательных) и сенсорных (чувствительных).
Если тип ШМТ демиелинизирующий, то разрушение или дефект миелиновой оболочки, которая защищает аксоны периферических нервов, приводит к замедлению передачи нервных импульсов периферической нервной системы – между мозгом, мышцами и органами чувств.
При аксональном типе болезни поражены непосредственно аксоны, что негативно отражается на силе нервных сигналов, которая недостаточна для полноценной стимуляции мышц и органов чувств.
Также читайте:
Как передается синдром Шарко-Мари-Тута? Дефектные гены могут быть унаследованы аутосомно-доминантным или аутосомно-рецессивным способами.
Наиболее частое – аутосомно-доминантное наследование – происходит при наличии одной копии мутировавшего гена (который несет один из родителей). И вероятность передачи ШМТ каждому из рожденных детей оценивается в 50%. [4]
При аутосомно-рецессивном наследовании для развития болезни нужны две копии дефектного гена (по одной от каждого родителя, у которых нет никаких признаков данного заболевания).
В 40-50% случаев происходит аутосомно-доминантное наследственная демиелинизации, то есть ШМТ 1 типа; в 12-26% случаев – ШМТ аксонального, то есть 2 типа. А в 10-15% случаев наблюдается Х-сцепленное наследование. [5]
Симптомы болезни Шарко-Мари-Тута
Обычно первые признаки данного заболевания начинают проявляются в детстве и у подростков и постепенно развиваются в течение всей жизни, хотя синдром может дать знать о себе и позже. Сочетание симптомов вариабельно, и скорость прогрессирования болезни, как и степень ее тяжести предсказать невозможно.
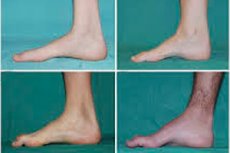
Отмечаются такие типичные симптомы начальной стадии, как повышенная общая утомляемость; снижение тонуса (слабость) мышц стоп, лодыжек и голеней; отсутствие рефлексов. Это затрудняет движение стопы и приводит к дисбазии (нарушению походки) в виде более высокого поднятия ног, нередко – с частыми спотыканиями и падениями. Признаками болезни Шарко-Мари-Тута у маленького ребенка может быть ярко выраженная неуклюжесть и несвойственные возрасту трудности при ходьбе, связанные с двусторонней свисающей стопой. Также характерны деформации стопы: высокий свод (полая стопа) или сильное плоскостопие, искривленные (молоткообразные) пальцы.
В случае ходьбы на пальцах ног на фоне мышечной гипотонии невролог может заподозрить у ребенка ШМТ 4 типа, при котором дети к подростковому возрасту могут быть не в состоянии ходить.
По мере прогрессирования мышечная атрофия и слабость распространяются на верхние конечности, затрудняя мелкую моторику и выполнение обычных действий руками. Снижение тактильных ощущений и способности чувствовать тепло и холод, а также онемение в стопах и кистях свидетельствует о повреждении аксонов сенсорных нервов.
При манифестирующей в детстве болезни Шарко-Мари-Тута 3 и 6 типов отмечается сенситивная атаксия (нарушение координации движений и равновесия), подергивание мышц и тремор, поражение лицевого нерва, атрофия зрительного нерва с нистагмом, снижение слуха.
На более поздних стадиях может быть неконтролируемая дрожь (тремор) и частые мышечные судороги; проблемы с передвижением могут привести к развитию боли: мышечной, суставной, невропатической.
Осложнения и последствия
Болезнь Шарко-Мари-Тута может иметь такие осложнения и последствия, как:
- более частые растяжения и переломы;
- контрактуры, связанные с укорочением околосуставных мышц и сухожилий;
- сколиоз (искривление позвоночника);
- проблемы с дыханием – при поражении нервных волокон, иннервирующих мышцы диафрагмы:
- утрата способности самостоятельно передвигаться.
Диагностика болезни Шарко-Мари-Тута
Диагностика включает клинический осмотр, анамнез (в том числе семейный), неврологическое и системное обследование.
Проводятся тесты для проверки диапазона движений, чувствительности и сухожильных рефлексов. Нервную проводимость позволяет оценить инструментальная диагностика – электромиография или электронейромиография. Также может потребоваться УЗИ или МРТ. [6]
Генетическая или ДНК диагностика для выявления при исследовании образца крови наиболее распространенных генетических мутаций, вызывающих ШМТ, ограничена, так как в настоящее время тесты ДНК доступны не для всех типов данного заболевания. Подробнее см. – Генетическое исследование
В некоторых случаях прибегают к биопсии периферического нерва (обычно икроножного).
Дифференциальная диагностика
Дифференциальная диагностика проводится с другими периферическими нейропатиями, мышечной дистрофией Дюшенна, миелопатическим и миастеническим синдромами, диабетической нейропатией, с миелоаптиями при рассеянном и боковом амиотрофическом склерозе, синдромом Гийена-Барре, травмой перонеального нерва и его атрофией (в том числе, при защемлении между поясничными дисками позвоночника), повреждением мозжечка или таламуса, а также с побочным действием химиотерапии (при лечении такими цитостатиками, как Винкристин или Паклитаксел). [7]
К кому обратиться?
Лечение болезни Шарко-Мари-Тута
На сегодняшний день лечение данного наследственного заболевания заключается в лечебной физкультуре (направленной на укрепление и растяжение мышц); трудотерапии (которая помогает пациентам с мышечной слабостью кистей); использовании ортопедических приспособлений для облегчения ходьбы. При необходимости назначается прием обезболивающих или противосудорожных препаратов. [8]
В случаях выраженного плоскостопия может проводиться остеотомия, а при деформации пяток показана их хирургическая коррекция – артродез. [9]
Ведутся исследования как генетической составляющей болезни, так и способов ее лечения. Применение стволовых клеток, некоторых гормонов, лецитина или аскорбиновой кислота пока не дали положительных результатов.
Но благодаря последним изысканиям в ближайшее время действительно может появиться новое в лечении болезни Шарко-Мари-Тута. Так, с 2014 года французской компанией Pharnext ведется разработка, а с середины 2019 года проходят клинические испытания препарата PXT3003 для лечения ШМТ 1 типа у взрослых, подавляющего повышенную экспрессию гена PMP22, улучшающего миелинизацию периферических нервов и ослабляющего нервно-мышечные симптомы.
Специалисты медицинской компании Sarepta Therapeutics (США) работают над созданием генной терапии 1 типа болезни Шарко-Мари-Тута. В этой терапии будет использоваться безвредный аденоассоциированный вирус (AAV) рода Dependovirus с линейным одноцепочечным ДНК-геномом, который будет переносить в организм ген NTF3, кодирующий необходимый для функционирования шванновских клеток нервов белок нейротрофин-3 (NT-3).
До конца 2020 года компания Helixmith приступит к клиническим испытаниям разработанной в Южной Корее генной терапии Engensis (VM202) – для лечения мышечных симптомов при ШМТ 1 типа. [10]
Профилактика
Профилактикой ШМТ может быть генетическая консультация будущих родителей, особенно, если кто-то из супружеской пары имеет в роду данное заболевание. Однако выявлены случаи точечных генных мутаций de novo, то есть при отсутствии болезни в семейном анамнезе.
Во время беременности проверить вероятность болезни Шарко-Мари-Тута у будущего ребенка позволяет биопсия ворсин хориона (с 10 по 13 неделю гестации), а также анализ околоплодных вод (на сроке 15-18 недель).
Прогноз
В целом, прогноз для разных типов болезни Шарко-Мари-Тута зависит от клинической тяжести, но в любом случае данное заболевание медленно прогрессирует. Многие пациенты имеют инвалидность, хотя это не снижает продолжительности жизни.