Медицинский эксперт статьи

Новые публикации
Алкаптонурия – врожденная ферментативная патология
Последняя редакция: 28.11.2021

Весь контент Web2Health проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Одно из очень редких нарушений обмена веществ – алкаптонурия – относится к врожденным аномалиям метаболизма аминокислоты тирозина.
Также данный синдром может называться дефицитом гомогентизатоксидазы, гомогентизинурией, наследственным охронозом или болезнью черной мочи. [1]
Код по МКБ-10
Эпидемиология
По статистике, на 1 млн. населения приходится не более девяти случаев алкаптонурии. А в большинстве стран Европы – один случай на 100-250 тыс. живорожденных младенцев.
Среди европейских стран исключением является Словакия (особенного относительно небольшой по территории северо-западный регион), где распространенность алкаптонурии составляет один случай на 19 тыс. новорожденных. По всей вероятности, это связано с тем, что среди проживающих там семей словацких ромов уровень инбридинга (браков между двоюродными родственниками) самый высокий в Европе: 10-14%. [2]
Причины алкаптонурии
Точные причины алкаптонурии, как врожденного расстройства катаболизма (метаболического распада) ароматической (гомоциклической) α-аминокислоты тирозина, установлены: данный тип нарушения обменных процессов является следствием гомозиготных или сложных гетерозиготных мутаций одного из тысячи генов на хромосоме 3, точнее – гена HGD в локусе 3q21-q23 на длинном плече хромосомы. Данным геном кодируются нуклеотидные последовательности фермента печени гомогентизат-1,2-диоксигеназы [3] (также называемого оксидазой гомогентизиновой кислоты или гомогентизатоксидазой) – железосодержащего металлопротеина, необходимого для одного из этапов расщепления тирозина в организме. [4], [5]
Таким образом, алкаптонурия – дефект фермента гомогентизат-1,2-диоксигеназы, точнее, результат его генетически обусловленного дефицита или полного отсутствия. [6]
Представляя собой врожденную ферментопатию, алкаптонурия наследуется как аутосомный рецессивный признак, то есть, чтобы возникла алкаптонурия у детей, должен быть видоизмененный ген указанного фермента у обоих родителей, поскольку каждый из них передает ребенку только одну копию гена из имеющихся двух.
По последним данным, насчитывается более двухсот вариантов видоизменения гена HGD, и чаще всего наблюдаются миссенс-мутации, транслокация и сплайсинг.
Факторы риска
Единственный фактор риска развития данной врожденной ферментопатии – наличие ее в семейном анамнезе и наследование двух видоизмененных копий гена HGD, если у родителей алкаптонурия не проявляется (риск передачи аномалии – 25%), или один из родителей имеет данное расстройство. [7]
Патогенез
Важнейшую роль тирозин играет в синтезе протеинов, выработке хромопротеинов – кожного пигмента меланина, а также тиреоидных гормонов и катехоламиновых нейротрансмиттеров.
Механизм регулирования количества тирозина в клетках очень сложный, и его избыточное содержание организм нормализует путем расщепления. Процесс катаболизма тирозина, как и всех ароматических аминокислот, многоступенчатый и протекает в несколько стадий. При этом каждый этап метаболического распада тирозина проходит с участием определенного фермента и образованием промежуточного соединения.
Так, сначала аминокислота расщепляется до пара-гидроксифенилпирувата, который превращается в алкаптон – 2,5-дигидроксифенилуксусную или гомогентизиновую кислоту. Далее алкаптон должен трансформироваться в малеуксусную кислоту, но этого не происходит. [8]
И патогенез алкаптонурии заключается в остановке биохимических реакций катаболизма тирозина на этапе образования гомогентизиновой кислоты: для ее расщепления просто нет нужного фермента – гомогентизатоксидазы.
Гомогентизиновая кислота организмом не используется и может накапливаться с выведением через почки. Кроме того, происходит ее окисление до бензохиноацетата (бензохинонуксусной кислоты), который, связываясь с молекулами тканей и жидкостей организма, образует биополимерные соединения, окрашенные подобно меланину.
Накопление в ткани данных промежуточных продуктов приводит к нарушению структуры коллагена хрящевой ткани, из-за чего снижается ее эластичность – с появлением многих клинических признаков алкаптонурии и развитием осложнений.
Симптомы алкаптонурии
Алкаптонурия у новорожденных и детей грудного возраста проявляется потемнением мочи. При контакте с воздухом цвет мочи на подгузниках, пеленках и белье становится темно-коричневым; это связано с накоплением и выделением гомогентизиновой кислоты, которая окисляется до бензохиноацетата. [10]
При отсутствии других симптомов нередко алкаптонурия у детей в раннем возрасте своевременно не распознается, так как после мочеиспускания моча может потемнеть через несколько часов. По некоторым данным, в условиях клиник выявляется лишь пятая часть детей до 12 месяцев, которые родились с данной ферментопатией. Поэтому очень важно внимание родителей к уходу за младенцем.
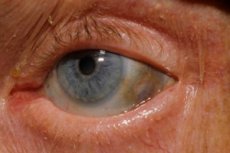
Кроме того, первые признаки включают пигментацию (синевато-серого цвета) глазных склер и хрящей ушных раковин и носа, которую часто называют охронозом. [11]
Со временем появляются и другие симптомы:
- сильная пигментация кожи на скулах, подмышечных впадин и гениталий;
- окрашивание одежды при контакте с потеющими участками тела;
- приступы общей слабости;
- хриплый голос.
Следует иметь в виду, что алкаптонурия и охроноз, как уже отмечалось выше, являются синонимичными названиями одного и того же нарушения катаболизма тирозина.
Болезнь кленового сиропа и алкаптонурия. Врожденная болезнь кленового сиропа или лейциноз тоже относится к метаболическим нарушениям, имеет тот же тип наследования, и даже мутации происходят на той же хромосоме, но касаются гена, кодирующего ферментный комплекс дегидрогеназы разветвленных α-кетокислот. Из-за этого организм не может расщеплять определенные компоненты белков, в частности, аминокислоты лейцин, изолейцин и валин. При данном заболевании моча (и ушная сера) имеет сладкий запах; кроме того, в клинической картине этого типа органической ацидемии наблюдаются гипопигментация, колебания АД, судороги, рвота и диарея, падение уровня глюкозы в крови, кетоацидоз, галлюцинации и др. Уровень смертности у детей достаточно высокий; у взрослых без лечения может наступить кома и летальный исход вследствие отека мозга.
Альбинизм и алкаптонурия «объединяет» только тирозин. Альбинизм, в том числе и окулокутанный, вызывается генетическими мутациями, которые влияют на выработку пигмента меланина. Врожденные изменения отмечаются в гене TYR на хромосоме 11 (11q14.3), который кодирует тирозиназу – содержащий медь фермент меланосом, необходимый для образования кожного пигмента на основе продуктов метаболизма тирозина. Данное заболевание намного более распространенное, чем алкаптонурия.
Осложнения и последствия
Обусловленные действием промежуточных метаболитов тирозина – гомогентизиновой и бензохинонуксусной кислот – последствия и осложнения алкаптонурии появляются из-за осаждения реактивных пигментированных полимеров, разрушения фибрилл коллагена и ухудшения состояния хрящей (со снижением их устойчивости к механическим нагрузкам).
Спустя годы, в зрелом возрасте, развивается дегенеративный артрит и остеоартрит крупных суставов (тазобедренных, крестцово-подвздошных и коленных); сужаются межпозвонковые пространства (особенно поясничного и грудного отделов позвоночника) – с кальцификацией и образованием остеофитов; снижается плотность ткани субхондральных костных пластин, а подлежащие кости могут подвергаться патологическому ремоделированию с образованием наростов и деформацией. [12]
Может наблюдаться повреждение сердечных клапанов (аортальных и митральных) и коронарных артерий – с признаками ишемической болезни сердца, а также образование камней в почках и предстательной железе – вследствие того же кальциноза. [13], [14]
Диагностика алкаптонурии
Обычно диагностика врожденных нарушений обмена веществ основана на исследовании биологических жидкостей организма.
На основе каких анализов и какими реакциями можно диагностировать алкаптонурию? Необходимы анализы мочи – для в обнаружения гомогентизиновой кислоты и определения ее уровня (нормальный – 20-30 мг в сутки, повышенный – 3-8 г). Образец мочи исследуется методом газовой хроматографии или масс-спектрометрии, с использованием жидкостной хроматографии; возможен скрининговый анализ на наличие хлорида железа в моче. [15]
Также существует метод быстрой диагностики – определение алкаптона в пятнах высохшей мочи на бумаге (по интенсивности цвета).
При уточнении диагноза инструментальная диагностика (рентгенография) предполагает выявление у пациентов рентгенологических признаков остеоартрита и других суставных патологий.
Подтверждают диагноз такие молекулярно-генетические методы диагностика наследственных заболеваний, как генетическое тестирование и секвенирование ДНК. [16]
Дифференциальная диагностика
Дифференциальная диагностика включает гемохроматоз и острую печеночную недостаточность новорожденных, меланинурию, острую перемежающуюся порфирию, гемофагоцитарный лимфогистиоцитоз, первичную митохондриальную патологию, ревматоидный артрит, анкилозирующий спондилит.
К кому обратиться?
Лечение алкаптонурии
Основное лечение алкаптонурии заключается в приеме внутрь больших доз (в день не менее 1000 мг) аскорбиновой кислоты. У детей это повышает выведение гомогентизиновой кислоты с мочой, а у взрослых снижает уровень содержания в моче ее деривата – бензохинонуксусной кислоты – и замедляет ее связывание с соединительнотканными структурами суставов и коллагеном. [17]
В западноевропейских клиниках проводят испытания лекарства Нитизинон (Орфалин) – препарата группы метаболиков, который ингибирует второй этап катаболизма тирозина: трансформацию пара-гидроксифенилпирувата в гомогентизиновую кислоту. Однако применение данного фармакологического средства приводит к накоплению тирозина и может вызывать тяжелые побочные эффекты, включая помутнение роговицы и светобоязнь, носовые и желудочные кровотечения, недостаточность печени, изменения со стороны крови и др. Тем не менее, в США Нитизинон одобрен FDA для лечения тирозинемии I типа. [18], [19]
Как таковое, физиотерапевтическое лечение – ЛФК для увеличения мышечной силы и повышения подвижности суставов, бальнео и пелоидоттерапия для ограничения боли – проводится при обусловленных алкаптонурией проблемах с суставами.
Хотя тирозин не только поступает с пищей, но и вырабатывается в организме, пациентам с алкаптонурией рекомендована диета с низким содержанием белка и ограничением потребления продуктов, богатых тирозином, в первую очередь, говядины и свинины, молочных продуктов (особенно сыров), бобовых, орехов и семян.
Профилактика
Профилактика генных мутаций невозможна, а для предупреждения появления на свет детей с высоким риском врожденных нарушений существует медико-генетическое консультирование, которое необходимо перед запланированной беременностью тех супружеских пар, в чьем семейном анамнезе имеются заболевания наследственного характера. [20]
Прогноз
Случаев фатального исхода течения алкаптонурии очень мало, и смерть может быть вызвана серьезными осложнениями, затрагивающими сердце и почки. Так что, для общей продолжительности жизни людей с алкаптонурией прогноз благоприятный.
Но качество жизни снижается из-за интенсивных болей в суставах или позвоночнике со значительным ограничением подвижности, часто прогрессирующим.